How to run Structure in HPC
 Structure_graph
Structure_graphJust give me credit whenever you get a chance 😃
This is a how to do file that I created for anyone in need to run STRUCTURE!! This is one of those programs that many people in the population genetics world use to determine genetic membership of populations. This tool was very important for me when I was doing my masters. Now that I have improved my understanding of bioinformatics and popgen I wanted to optimize the way this is run.
STRUCTURE is program that runs in serial. To parallelize/optimize structure, Pina-Martins et.al created a program called structure_threader. See more info about it here.
Table of Contents
Before you start:
- You need access to a High Performance Computer(HPC). The context of this turorial is around the resources available at NC State, Henry2 is the name of the HPC that I have access to. The installation may vary depending of what kind of machine you are running.
- You need to have an input file! Needs to be in structure format which is very particular. See here. If you do not have this format available, you can always convert what you have.
- You need to know some basic information about your system: Plody level, number of individuals and number of loci.
1) Install STRUCTURE in HPC
In order to install programs in the HPC, you need acces to the usrapps/ directory if you do not have it. Please request it via email to your hpc help email.
Open the terminal:
ssh unityID@login.hpc.ncsu.edu #Sing in with your password and duo authentification.
Once inside, reset any previous environment used by typing:
module purge
Then load conda:
module load conda
conda init tcsh
Now log out by typing:
exit
Then log back in and you only need to do module and conda commands once:
ssh unityID@login.hpc.ncsu.edu
Lets now create a .condarc in your home directory that tells conda NOT to put downloaded source files to your home directory, (e.g. put it in share) containing the following:
Use vi:
cd ~ #make sure to be located in home
vi .condarc
You just type i to edit and the copy and paste from here:
pkgs_dirs:
- /share/$GROUP/$USER/conda/pkgs
To save the changes and to close type ESC followed by Shift ZZ
Now lets create the environment (make sure to chage PI_unitID to your PI’s unityID), do:
conda create --prefix /usr/local/usrapps/PI_unityID/env_structure_threader
conda activate /usr/local/usrapps/PI_unityID/env_structure_threader
conda install pip
pip install structure_threader
Now deactivate the enviroment, do:
conda deactivate
And everytime you run again, you don’t have to do module load or source the file because conda init puts that in your path. The only thing you need to do is to add the following to your jobs before running structure_threader:
conda activate /usr/local/usrapps/PI_unityID/env_structure_threader
2) Prepare mainparams file
To get structure to work with your parameters, you need to modify the mainparams. I would say that at this point you need to read the manual to determine change in the file.
Lets first go to the location where structure will be lauched:
cd /share/PI_unityID/YOUR_unityID/
mkdir structure #Create a directory to put
To modify this file use vi (command line text editor):
vi mainparams
This opens the file, now to make chages type i and go a head and edit this file as you
KEY PARAMETERS FOR THE PROGRAM structure. YOU WILL NEED TO SET THESE
IN ORDER TO RUN THE PROGRAM. VARIOUS OPTIONS CAN BE ADJUSTED IN THE
FILE extraparams.
"(int)" means that this takes an integer value.
"(B)" means that this variable is Boolean
(ie insert 1 for True, and 0 for False)
"(str)" means that this is a string (but not enclosed in quotes!)
Basic Program Parameters
#define MAXPOPS 20 // (int) number of populations assumed
#define BURNIN 100000 // (int) length of burnin period
#define NUMREPS 1000000 // (int) number of MCMC reps after burnin
Input/Output files
#define INFILE infile // (str) name of input data file
#define OUTFILE outfile //(str) name of output data file
Data file format
#define NUMINDS 134 // (int) number of diploid individuals in data file
#define NUMLOCI 12 // (int) number of loci in data file
#define PLOIDY 2 // (int) ploidy of data
#define MISSING 0 // (int) value given to missing genotype data
#define ONEROWPERIND 0 // (B) store data for individuals in a single line
#define LABEL 1 // (B) Input file contains individual labels
#define POPDATA 1 // (B) Input file contains a population identifier
#define POPFLAG 0 // (B) Input file contains a flag which says
whether to use popinfo when USEPOPINFO==1
#define LOCDATA 0 // (B) Input file contains a location identifier
#define PHENOTYPE 0 // (B) Input file contains phenotype information
#define EXTRACOLS 0 // (int) Number of additional columns of data
before the genotype data start.
#define MARKERNAMES 1 // (B) data file contains row of marker names
#define RECESSIVEALLELES 0 // (B) data file contains dominant markers (eg AFLPs)
// and a row to indicate which alleles are recessive
#define MAPDISTANCES 0 // (B) data file contains row of map distances
// between loci
Advanced data file options
#define PHASED 0 // (B) Data are in correct phase (relevant for linkage model only)
#define PHASEINFO 0 // (B) the data for each individual contains a line
indicating phase (linkage model)
#define MARKOVPHASE 0 // (B) the phase info follows a Markov model.
#define NOTAMBIGUOUS -999 // (int) for use in some analyses of polyploid data
Command line options:
-m mainparams
-e extraparams
-s stratparams
-K MAXPOPS
-L NUMLOCI
-N NUMINDS
-i input file
-o output file
-D SEED
To save the changes and to close type ESC followed by Shift ZZ
3) Prepare extraparams file
We also need to create an extraparams file that structure uses. Sometimes will be necessary to modify the extraparams file to run structure. See bellow the options to modify:
vi extraparams
Now make the changes:
EXTRA PARAMS FOR THE PROGRAM structure. THESE PARAMETERS CONTROL HOW THE
PROGRAM RUNS. ATTRIBUTES OF THE DATAFILE AS WELL AS K AND RUNLENGTH ARE
SPECIFIED IN mainparams.
"(int)" means that this takes an integer value.
"(d)" means that this is a double (ie, a Real number such as 3.14).
"(B)" means that this variable is Boolean
(ie insert 1 for True, and 0 for False).
PROGRAM OPTIONS
#define NOADMIX 0 // (B) Use no admixture model (0=admixture model, 1=no-admix)
#define LINKAGE 0 // (B) Use the linkage model model
#define USEPOPINFO 0 // (B) Use prior population information to pre-assign individuals
to clusters
#define LOCPRIOR 0 //(B) Use location information to improve weak data
#define FREQSCORR 1 // (B) allele frequencies are correlated among pops
#define ONEFST 0 // (B) assume same value of Fst for all subpopulations.
#define INFERALPHA 1 // (B) Infer ALPHA (the admixture parameter)
#define POPALPHAS 0 // (B) Individual alpha for each population
#define ALPHA 1.0 // (d) Dirichlet parameter for degree of admixture
(this is the initial value if INFERALPHA==1).
#define INFERLAMBDA 0 // (B) Infer LAMBDA (the allele frequencies parameter)
#define POPSPECIFICLAMBDA 0 //(B) infer a separate lambda for each pop
(only if INFERLAMBDA=1).
#define LAMBDA 1.0 // (d) Dirichlet parameter for allele frequencies
PRIORS
#define FPRIORMEAN 0.01 // (d) Prior mean and SD of Fst for pops.
#define FPRIORSD 0.05 // (d) The prior is a Gamma distribution with these parameters
#define UNIFPRIORALPHA 1 // (B) use a uniform prior for alpha;
otherwise gamma prior
#define ALPHAMAX 10.0 // (d) max value of alpha if uniform prior
#define ALPHAPRIORA 1.0 // (only if UNIFPRIORALPHA==0): alpha has a gamma
prior with mean A*B, and
#define ALPHAPRIORB 2.0 // variance A*B^2.
#define LOG10RMIN -4.0 //(d) Log10 of minimum allowed value of r under linkage model
#define LOG10RMAX 1.0 //(d) Log10 of maximum allowed value of r
#define LOG10RPROPSD 0.1 //(d) standard deviation of log r in update
#define LOG10RSTART -2.0 //(d) initial value of log10 r
USING PRIOR POPULATION INFO (USEPOPINFO)
#define GENSBACK 2 //(int) For use when inferring whether an indiv-
idual is an immigrant, or has an immigrant an-
cestor in the past GENSBACK generations. eg, if
GENSBACK==2, it tests for immigrant ancestry
back to grandparents.
#define MIGRPRIOR 0.01 //(d) prior prob that an individual is a migrant
(used only when USEPOPINFO==1). This should
be small, eg 0.01 or 0.1.
#define PFROMPOPFLAGONLY 0 // (B) only use individuals with POPFLAG=1 to update P.
This is to enable use of a reference set of
individuals for clustering additional "test"
individuals.
LOCPRIOR MODEL FOR USING LOCATION INFORMATION
#define LOCISPOP 1 //(B) use POPDATA for location information
#define LOCPRIORINIT 1.0 //(d) initial value for r, the location prior
#define MAXLOCPRIOR 20.0 //(d) max allowed value for r
OUTPUT OPTIONS
#define PRINTNET 1 // (B) Print the "net nucleotide distance" to screen during the run
#define PRINTLAMBDA 1 // (B) Print current value(s) of lambda to screen
#define PRINTQSUM 1 // (B) Print summary of current population membership to screen
#define SITEBYSITE 0 // (B) whether or not to print site by site results.
(Linkage model only) This is a large file!
#define PRINTQHAT 0 // (B) Q-hat printed to a separate file. Turn this
on before using STRAT.
#define UPDATEFREQ 100 // (int) frequency of printing update on the screen.
Set automatically if this is 0.
#define PRINTLIKES 0 // (B) print current likelihood to screen every rep
#define INTERMEDSAVE 0 // (int) number of saves to file during run
#define ECHODATA 1 // (B) Print some of data file to screen to check
that the data entry is correct.
(NEXT 3 ARE FOR COLLECTING DISTRIBUTION OF Q:)
#define ANCESTDIST 0 // (B) collect data about the distribution of an-
cestry coefficients (Q) for each individual
#define NUMBOXES 1000 // (int) the distribution of Q values is stored as
a histogram with this number of boxes.
#define ANCESTPINT 0.90 // (d) the size of the displayed probability
interval on Q (values between 0.0--1.0)
MISCELLANEOUS
#define COMPUTEPROB 1 // (B) Estimate the probability of the Data under
the model. This is used when choosing the
best number of subpopulations.
#define ADMBURNIN 500 // (int) [only relevant for linkage model]:
Initial period of burnin with admixture model (see Readme)
#define ALPHAPROPSD 0.025 // (d) SD of proposal for updating alpha
#define STARTATPOPINFO 0 // Use given populations as the initial condition
for population origins. (Need POPDATA==1). It
is assumed that the PopData in the input file
are between 1 and k where k<=MAXPOPS.
#define RANDOMIZE 1 // (B) use new random seed for each run
#define SEED 2245 // (int) seed value for random number generator
(must set RANDOMIZE=0)
#define METROFREQ 10 // (int) Frequency of using Metropolis step to update
Q under admixture model (ie use the metr. move every
i steps). If this is set to 0, it is never used.
(Proposal for each q^(i) sampled from prior. The
goal is to improve mixing for small alpha.)
#define REPORTHITRATE 0 // (B) report hit rate if using METROFREQ
4) Prepare your input file
Structure is very particular about the format in which the individuals, alleles, loci, or any other info is organized. This is an example of an imput file:
A107 A29 AP273 AC306 AP55 A24 A88 B124 AP43 AP81 A113 AP66
KA133 1 168 0 108 162 0 102 136 213 131 126 211 92
KA133 1 168 0 108 182 0 108 140 222 140 126 217 98
KA143 1 0 0 108 162 176 102 136 213 131 126 211 92
KA143 1 0 0 108 162 176 108 140 222 140 134 217 98
KA144 1 162 152 108 162 176 102 136 213 131 126 205 92
KA144 1 174 152 108 176 176 108 140 222 140 134 217 92
Notice the first row corresponds to the loci.
Each individual has 2 rows since it is a diploid organism.
The first column is the individual label.
The second column provides population information in this case 1 represents individuals from the population #1.
In the center, you can see the actual alleles for the respective loci.
5)Prepare the hpc job
Now that we have our files:
- Input file
- mainparams
- extraparams
We can create an HPC job to run structure in parallel. Preparing an HPC job means creating a script that schedules resources in the HPC and directs the commands you actually want to run. This preparation can be accomplished using vi like we have done before, copy and paste the job bellow and make changes to fit your unityID:
#!/bin/csh
##Structure run N=134 and L=12
#BSUB -o out.%J
#BSUB -e err.%J
#BSUB -W 48:00
#BSUB -n 16
#BSUB -q shared_memory
#BSUB -R "rusage[mem=32000] span[hosts=1]"
#BSUB -J structure_N134_L12
conda activate /usr/local/usrapps/PI_unityID/env_structure_threader
structure_threader run -K 20 -R 5 -i /gpfs_common/share03/PI_unityID/YOUR_unityID/structure/input_structure_N134_L12.txt -o /gpfs_common/share03/PI_unityID/YOUR_unityID/
structure/ -t 16 -st /usr/local/usrapps/PI_unityID/env_structure_threader/bin/structure
This will run in about 1.72 hours!!! Which is amazing 😏 –> 🔓
6) Once your job is complete! move the results to your personal computer:
Use scp to move whole folders to your personal computer. Open a new terminal window and navegate to your desire location to copy the files then type:
#To move from HPC to personal computer:
scp -r YOUR_unityID@login.hpc.ncsu.edu:/share/PI_unityID/YOUR_unityID/structure .
#The . at the end means current directory in your personal computer!!
7) Determine your bestK using structure harvester:
Now that you have your files in your personal computer, you can determine the best K for your data. In the example below, the bestK is 3.
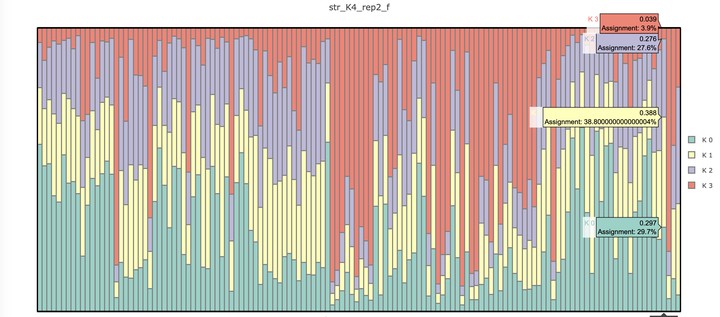
8) Create structure plots!
The last step is to create those famous plots. Structure_threader creates some default plots which can be all you need but if you want to try to disect the information, you will have to create the plot in something like excel or R.
This can be done using one of the files for the K that you determined to be your best K. The membership section of that file will contain K membership proportions for each individual.
Inferred ancestry of individuals:
Label (%Miss) Pop: Inferred clusters
1 KA133 (16) 1 : 0.109 0.079 0.812
2 KA143 (16) 1 : 0.124 0.155 0.721
3 KA144 (0) 1 : 0.101 0.106 0.793
4 KA145 (0) 1 : 0.305 0.090 0.605
5 KA146 (8) 1 : 0.365 0.107 0.528
6 KA147 (16) 1 : 0.180 0.063 0.757
7 KA148 (0) 1 : 0.222 0.060 0.719
8 KA61 (0) 1 : 0.128 0.129 0.743
9 KA62 (0) 1 : 0.306 0.047 0.647
10 KA63 (0) 1 : 0.258 0.454 0.288
These table can be move to excell or R to make the stack graph.